Tidyverse and the OMOP CDM
This guide introduces a powerful and intuitive approach to working with OMOP CDM data using R and the tidyverse. The tidyverse is a collection of R packages designed for data science that share a common design philosophy, grammar, and data structures. By leveraging these tools, you can write clean, readable, and efficient code to perform complex analyses on large-scale observational health data.
Who is this guide for?
This guide is written for anyone who wants to work with databases in a “tidyverse” style—a human-centered, consistent, and composable approach to data analysis.
- New to R? We recommend complementing this guide with R for Data Science.
- New to databases? Familiarize yourself with SQL basics through tutorials like SQLBolt or SQLZoo.
- New to the OMOP CDM? This guide is best paired with The Book of OHDSI.
Bridging the Gap for Different Backgrounds
This guide is designed to be accessible to readers from various backgrounds. Here’s how we address common challenges:
For SAS Programmers
If you’re coming from SAS, you might find the functional programming style of R unfamiliar. Here’s a quick mapping of common SAS concepts to their R/dplyr equivalents:
| SAS Concept | R/dplyr Equivalent | Description |
|---|---|---|
PROC SQL | dplyr verbs like filter(), select(), mutate() | Querying and manipulating data |
DATA step | mutate() or transmute() | Creating or modifying variables |
PROC SORT | arrange() | Sorting data |
PROC SUMMARY | group_by() + summarise() | Grouping and aggregating data |
PROC FREQ | count() | Frequency tables |
Remember, in R, operations are chained using the pipe (|>) for readability, similar to how you might chain procedures in SAS.
For Clinical Experts
If you’re new to data analysis but have a strong clinical background, the OMOP CDM might seem overly complex at first. Think of it as a standardized “language” for health data:
- Why separate tables? Instead of one big spreadsheet, data is split into logical tables (e.g.,
personfor demographics,condition_occurrencefor diagnoses) to avoid repetition and ensure consistency. - What are concept IDs? These are standardized codes for medical terms (e.g., “Type 2 Diabetes” might have ID 201826). They ensure everyone uses the same definitions, making research comparable.
- Why vocabularies? OMOP uses controlled vocabularies (like SNOMED or ICD) to map local codes to standard ones, reducing ambiguity.
This structure allows for powerful, scalable analyses across different healthcare systems.
For Data Scientists New to Healthcare
If you’re proficient in R but unfamiliar with clinical research, we’ll explain the “why” behind the code. For example, when we create a “cohort” of patients, it’s not just filtering data—it’s defining a study population based on clinical criteria to answer specific research questions.
How is this guide organized?
This guide is divided into two main parts:
- General Principles for Working with Databases in R: The first half focuses on the foundational concepts of using tidyverse-style code to build analytical pipelines. You will learn how to connect to a database, manipulate data, and prepare it for analysis without ever leaving the R environment.
- Applying these Principles to the OMOP CDM: The second half demonstrates how to apply these general principles specifically to the OMOP Common Data Model. We will explore a suite of specialized R packages that streamline common analytical tasks in observational health research.
Detailed Chapters Overview
For a deeper dive, the underlying technical manual for this guide is structured into the following detailed chapters. While this page provides a high-level summary, the full manual offers comprehensive examples and technical explanations.
1. A first analysis using data in a database
The iris dataset is a classic example in data science, collected by Edgar Anderson in 1935. It contains measurements of 150 iris flowers from three species: setosa, versicolor, and virginica. Each flower has four measurements: sepal length, sepal width, petal length, and petal width (all in centimeters). The goal is often to classify the species based on these measurements, making it a simple but effective dataset for demonstrating statistical and machine learning techniques.
Setting Up Your Environment
First, you need to install and load the necessary R packages.
# Load the libraries
library(dplyr)
library(dbplyr)
library(ggplot2)
library(DBI)
library(duckdb)
Inserting Data into a Database
Next, we will load the iris data into an in-memory duckdb database. This simulates a real-world scenario where your data resides in a database server.
# Create an in-memory duckdb database
db <- dbConnect(drv = duckdb())
# Write the iris dataframe to a table named "iris" in the database
dbWriteTable(db, "iris", iris)
# You can see the tables in the database
dbListTables(db)
## [1] "iris"
# > [1] "iris"
From R to SQL: The Power of dbplyr
Now that the data is in a database, we could query it using SQL. However, the magic of the dbplyr package is that it allows you to write familiar dplyr code, which it automatically translates into SQL for you.
First, create a reference to the iris table in the database.
iris_db <- tbl(db, "iris")
Now, you can use standard dplyr verbs on this object.
# Get a summary of sepal length by species
iris_db |>
group_by(Species) |>
summarise(
n = n(),
mean_sepal_length = mean(Sepal.Length, na.rm = TRUE)
)
## # Source: SQL [?? x 3]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Species n mean_sepal_length
## <fct> <dbl> <dbl>
## 1 setosa 50 5.01
## 2 versicolor 50 5.94
## 3 virginica 50 6.59
When you run this code, dbplyr translates it into the following SQL query, sends it to the database, and returns the result.
<SQL>
SELECT
"Species",
COUNT(*) AS n,
AVG("Sepal.Length") AS mean_sepal_length
FROM iris
GROUP BY "Species"
Bringing Data into R for Visualization
While most data manipulation should happen in the database for efficiency, you will often need to bring the final, summarized data back into R for visualization or modeling. The collect() function does this.
Let’s create a histogram of sepal length for each flower species.
iris_db |>
select("Species", "Sepal.Length") |>
collect() |> # This brings the data from the database into an R dataframe
ggplot(aes(x = `Sepal.Length`, fill = Species)) +
geom_histogram(binwidth = 0.2, colour = "black") +
facet_wrap(~Species, ncol = 1) +
theme_bw()
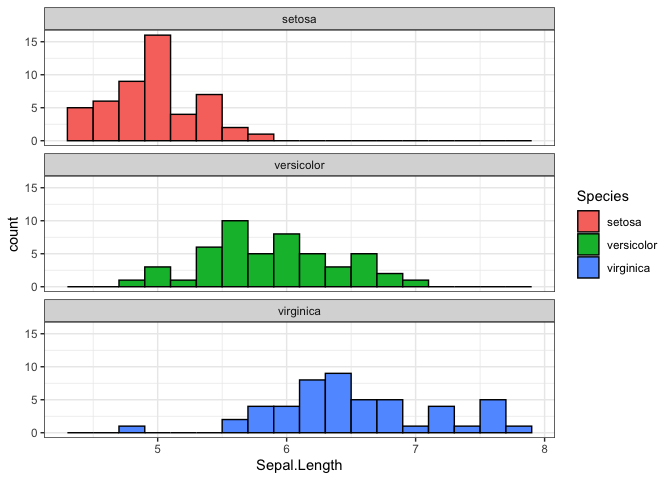
This workflow—manipulating data in the database and collecting only the results—is the most efficient way to work with large datasets.
Disconnecting from the Database
When you are finished with your analysis, it is good practice to close the connection to the database.
dbDisconnect(db)
2. Core verbs for analytic pipelines utilising a database
To demonstrate working with multiple tables, we’ll extend the iris example by creating related tables. We’ll split the data into separate tables for species information and measurements, then show how to join them back together.
Setting Up the Database with Multiple Tables
# Create database
db <- dbConnect(duckdb())
# Split iris into two tables: species info and measurements
species_info <- iris |> select(Species) |> distinct() |> mutate(species_id = row_number())
measurements <- iris |> left_join(species_info, by = "Species")
# Write tables
dbWriteTable(db, "species_info", species_info)
dbWriteTable(db, "measurements", measurements)
# Create lazy references
species_db <- tbl(db, "species_info")
measurements_db <- tbl(db, "measurements")
Selecting Rows: filter() and distinct()
Use filter() to select rows based on conditions. For example, to find measurements with sepal length > 5:
measurements_db |>
filter(Sepal.Length > 5) |>
select(Species, Sepal.Length)
## # Source: SQL [?? x 2]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Species Sepal.Length
## <fct> <dbl>
## 1 setosa 5.1
## 2 setosa 5.4
## 3 setosa 5.4
## 4 setosa 5.8
## 5 setosa 5.7
## 6 setosa 5.4
## 7 setosa 5.1
## 8 setosa 5.7
## 9 setosa 5.1
## 10 setosa 5.4
## # ℹ more rows
distinct() removes duplicate rows:
measurements_db |>
distinct(Species)
## # Source: SQL [?? x 1]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Species
## <fct>
## 1 setosa
## 2 versicolor
## 3 virginica
Ordering Rows: arrange()
arrange() sorts rows by one or more columns:
measurements_db |>
arrange(desc(Sepal.Length)) |>
select(Species, Sepal.Length)
## # Source: SQL [?? x 2]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## # Ordered by: desc(Sepal.Length)
## Species Sepal.Length
## <fct> <dbl>
## 1 virginica 7.9
## 2 virginica 7.7
## 3 virginica 7.7
## 4 virginica 7.7
## 5 virginica 7.7
## 6 virginica 7.6
## 7 virginica 7.4
## 8 virginica 7.3
## 9 virginica 7.2
## 10 virginica 7.2
## # ℹ more rows
Column Transformation: mutate(), select(), rename()
mutate() creates or modifies columns:
measurements_db |>
mutate(
sepal_ratio = Sepal.Length / Sepal.Width,
is_large = Sepal.Length > 6
) |>
select(Species, sepal_ratio, is_large)
## # Source: SQL [?? x 3]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Species sepal_ratio is_large
## <fct> <dbl> <lgl>
## 1 setosa 1.46 FALSE
## 2 setosa 1.63 FALSE
## 3 setosa 1.47 FALSE
## 4 setosa 1.48 FALSE
## 5 setosa 1.39 FALSE
## 6 setosa 1.38 FALSE
## 7 setosa 1.35 FALSE
## 8 setosa 1.47 FALSE
## 9 setosa 1.52 FALSE
## 10 setosa 1.58 FALSE
## # ℹ more rows
select() chooses specific columns:
measurements_db |>
select(Species, Sepal.Length, Petal.Length)
## # Source: SQL [?? x 3]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Species Sepal.Length Petal.Length
## <fct> <dbl> <dbl>
## 1 setosa 5.1 1.4
## 2 setosa 4.9 1.4
## 3 setosa 4.7 1.3
## 4 setosa 4.6 1.5
## 5 setosa 5 1.4
## 6 setosa 5.4 1.7
## 7 setosa 4.6 1.4
## 8 setosa 5 1.5
## 9 setosa 4.4 1.4
## 10 setosa 4.9 1.5
## # ℹ more rows
rename() changes column names:
measurements_db |>
rename(sepal_length = Sepal.Length)
## # Source: SQL [?? x 6]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## sepal_length Sepal.Width Petal.Length Petal.Width Species species_id
## <dbl> <dbl> <dbl> <dbl> <fct> <int>
## 1 5.1 3.5 1.4 0.2 setosa 1
## 2 4.9 3 1.4 0.2 setosa 1
## 3 4.7 3.2 1.3 0.2 setosa 1
## 4 4.6 3.1 1.5 0.2 setosa 1
## 5 5 3.6 1.4 0.2 setosa 1
## 6 5.4 3.9 1.7 0.4 setosa 1
## 7 4.6 3.4 1.4 0.3 setosa 1
## 8 5 3.4 1.5 0.2 setosa 1
## 9 4.4 2.9 1.4 0.2 setosa 1
## 10 4.9 3.1 1.5 0.1 setosa 1
## # ℹ more rows
Grouping and Aggregation: group_by(), summarise(), count()
group_by() groups data for calculations:
measurements_db |>
group_by(Species) |>
summarise(
avg_sepal = mean(Sepal.Length, na.rm = TRUE),
count = n()
)
## # Source: SQL [?? x 3]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Species avg_sepal count
## <fct> <dbl> <dbl>
## 1 setosa 5.01 50
## 2 versicolor 5.94 50
## 3 virginica 6.59 50
count() is a shortcut for counting:
measurements_db |>
count(Species, sort = TRUE)
## # Source: SQL [?? x 2]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## # Ordered by: desc(n)
## Species n
## <fct> <dbl>
## 1 setosa 50
## 2 versicolor 50
## 3 virginica 50
Joining Tables
Joins combine data from multiple tables. For example, to join measurements with species info:
measurements_db |>
left_join(species_db, by = "Species") |>
select(Species, Sepal.Length)
## # Source: SQL [?? x 2]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Species Sepal.Length
## <fct> <dbl>
## 1 setosa 5.1
## 2 setosa 4.9
## 3 setosa 4.7
## 4 setosa 4.6
## 5 setosa 5
## 6 setosa 5.4
## 7 setosa 4.6
## 8 setosa 5
## 9 setosa 4.4
## 10 setosa 4.9
## # ℹ more rows
Constructing a Tidy Analytic Dataset
Let’s create a comprehensive dataset by joining tables and performing aggregations:
analytic_dataset <- measurements_db |>
group_by(Species) |>
summarise(
avg_sepal_length = mean(Sepal.Length, na.rm = TRUE),
avg_petal_length = mean(Petal.Length, na.rm = TRUE),
total_measurements = n()
) |>
arrange(desc(avg_sepal_length))
# Collect for analysis
analytic_dataset |> collect()
## # A tibble: 3 × 4
## Species avg_sepal_length avg_petal_length total_measurements
## <fct> <dbl> <dbl> <dbl>
## 1 virginica 6.59 5.55 50
## 2 versicolor 5.94 4.26 50
## 3 setosa 5.01 1.46 50
This pipeline demonstrates how to build complex queries using dplyr verbs, with all operations pushed to the database for efficiency.
3. Supported expressions for database queries
Data Type Conversions
R and SQL handle data types differently. dbplyr automatically converts many types, but understanding the mappings is important:
- Logical:
TRUE/FALSE→TRUE/FALSEor1/0 - Character: Strings remain strings
- Numeric: Integers and doubles are preserved
- Dates:
Dateobjects →DATE - Datetimes:
POSIXct→TIMESTAMP
Logical Comparisons and Operators
Standard comparison operators work as expected:
measurements_db |> filter(Sepal.Length > 5 & Petal.Length > 3)
## # Source: SQL [?? x 6]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Sepal.Length Sepal.Width Petal.Length Petal.Width Species species_id
## <dbl> <dbl> <dbl> <dbl> <fct> <int>
## 1 7 3.2 4.7 1.4 versicolor 2
## 2 6.4 3.2 4.5 1.5 versicolor 2
## 3 6.9 3.1 4.9 1.5 versicolor 2
## 4 5.5 2.3 4 1.3 versicolor 2
## 5 6.5 2.8 4.6 1.5 versicolor 2
## 6 5.7 2.8 4.5 1.3 versicolor 2
## 7 6.3 3.3 4.7 1.6 versicolor 2
## 8 6.6 2.9 4.6 1.3 versicolor 2
## 9 5.2 2.7 3.9 1.4 versicolor 2
## 10 5.9 3 4.2 1.5 versicolor 2
## # ℹ more rows
# Translates to: WHERE "Sepal.Length" > 5 AND "Petal.Length" > 3
Conditional Statements
if_else() and case_when() are supported:
measurements_db |>
mutate(
size_category = case_when(
Sepal.Length < 5 ~ "small",
Sepal.Length <= 6 ~ "medium",
TRUE ~ "large"
)
)
## # Source: SQL [?? x 7]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Sepal.Length Sepal.Width Petal.Length Petal.Width Species species_id
## <dbl> <dbl> <dbl> <dbl> <fct> <int>
## 1 5.1 3.5 1.4 0.2 setosa 1
## 2 4.9 3 1.4 0.2 setosa 1
## 3 4.7 3.2 1.3 0.2 setosa 1
## 4 4.6 3.1 1.5 0.2 setosa 1
## 5 5 3.6 1.4 0.2 setosa 1
## 6 5.4 3.9 1.7 0.4 setosa 1
## 7 4.6 3.4 1.4 0.3 setosa 1
## 8 5 3.4 1.5 0.2 setosa 1
## 9 4.4 2.9 1.4 0.2 setosa 1
## 10 4.9 3.1 1.5 0.1 setosa 1
## # ℹ more rows
## # ℹ 1 more variable: size_category <chr>
SQL translation:
CASE
WHEN "Sepal.Length" < 5 THEN 'small'
WHEN "Sepal.Length" <= 6 THEN 'medium'
ELSE 'large'
END
String Functions
Common string operations:
species_db |>
mutate(
species_upper = toupper(Species),
species_length = nchar(Species),
species_substr = substr(Species, 1, 3)
)
## # Source: SQL [?? x 5]
## # Database: DuckDB 1.4.0 [root@Darwin 25.1.0:R 4.5.2/:memory:]
## Species species_id species_upper species_length species_substr
## <fct> <int> <chr> <dbl> <chr>
## 1 setosa 1 SETOSA 6 set
## 2 versicolor 2 VERSICOLOR 10 ver
## 3 virginica 3 VIRGINICA 9 vir
Database-Specific Considerations
While dbplyr aims for portability, some functions may behave differently:
- DuckDB: Full support for most R functions
- PostgreSQL: Excellent date/time support
- SQL Server: May require workarounds for some functions
Always test your queries on your target database system.
4. Building analytic pipelines for a data model
Principles of Modular Pipelines
- Separation of Concerns: Break down complex queries into smaller, focused steps
- Reusability: Create functions for common operations
- Readability: Use clear variable names and comments
- Efficiency: Minimize data movement between database and R
Example: Analyzing Iris Measurements
Building on the iris example, let’s create a pipeline to analyze measurements by species:
# Step 1: Filter and prepare data
iris_performance <- measurements_db |>
filter(!is.na(Sepal.Length), !is.na(Petal.Length))
# Step 2: Calculate metrics
performance_metrics <- iris_performance |>
mutate(
sepal_petal_ratio = Sepal.Length / Petal.Length,
is_large_flower = as.numeric(Sepal.Length > 6)
) |>
group_by(Species) |>
summarise(
total_flowers = n(),
avg_sepal_petal_ratio = mean(sepal_petal_ratio, na.rm = TRUE),
large_percentage = mean(is_large_flower, na.rm = TRUE) * 100
)
# Step 3: Filter for significant groups
significant_species <- performance_metrics |>
filter(total_flowers >= 40) |>
arrange(desc(avg_sepal_petal_ratio))
# Collect results
results <- significant_species |> collect()
Creating Reusable Functions
For repeated operations, create functions:
calculate_ratios <- function(measurements_tbl) {
measurements_tbl |>
mutate(
sepal_petal_ratio = Sepal.Length / Petal.Length,
is_large = as.numeric(Sepal.Length > 6)
) |>
summarise(
total = n(),
avg_ratio = mean(sepal_petal_ratio, na.rm = TRUE),
large_pct = mean(is_large, na.rm = TRUE)
)
}
# Use the function
species_ratios <- measurements_db |>
group_by(Species) |>
calculate_ratios()
Handling Complex Joins in Relational Models
In structured data models like OMOP, you’ll often need to join multiple tables. Plan your joins carefully:
- Identify the central table (e.g.,
personin OMOP) - Determine the join keys
- Consider the join type (left, inner, etc.)
- Chain joins logically
This approach ensures your pipelines are maintainable and efficient.
Part 2: Applying these Principles to the OMOP CDM
5. Creating a CDM reference
library(CDMConnector)
library(omock)
library(lubridate)
Connecting to a Local OMOP Database
For a local DuckDB file:
cdm <- cdmFromCon(
con = dbConnect(duckdb(), "path/to/omop.db"),
cdmSchema = "main",
writeSchema = "main"
)
Using Mock Data for Learning
cdm <- mockCdmReference()
This creates a cdm object with sample OMOP data.
Exploring the CDM Object
# List available tables
names(cdm)
## [1] "person" "observation_period" "cdm_source"
## [4] "concept" "vocabulary" "concept_relationship"
## [7] "concept_synonym" "concept_ancestor" "drug_strength"
# Access specific tables
cdm$person
## # A tibble: 0 × 18
## # ℹ 18 variables: person_id <int>, gender_concept_id <int>,
## # year_of_birth <int>, month_of_birth <int>, day_of_birth <int>,
## # birth_datetime <date>, race_concept_id <int>, ethnicity_concept_id <int>,
## # location_id <int>, provider_id <int>, care_site_id <int>,
## # person_source_value <chr>, gender_source_value <chr>,
## # gender_source_concept_id <int>, race_source_value <chr>,
## # race_source_concept_id <int>, ethnicity_source_value <chr>, …
Understanding the CDM Structure
The cdm object knows the relationships between tables:
cdm$person: Patient demographicscdm$condition_occurrence: Diagnosescdm$drug_exposure: Medicationscdm$visit_occurrence: Healthcare visits
Verifying Connection
# Count patients
cdm$person |> count() |> collect()
## # A tibble: 1 × 1
## n
## <int>
## 1 0
# Check table structure
cdm$person |> glimpse()
## Rows: 0
## Columns: 18
## $ person_id <int>
## $ gender_concept_id <int>
## $ year_of_birth <int>
## $ month_of_birth <int>
## $ day_of_birth <int>
## $ birth_datetime <date>
## $ race_concept_id <int>
## $ ethnicity_concept_id <int>
## $ location_id <int>
## $ provider_id <int>
## $ care_site_id <int>
## $ person_source_value <chr>
## $ gender_source_value <chr>
## $ gender_source_concept_id <int>
## $ race_source_value <chr>
## $ race_source_concept_id <int>
## $ ethnicity_source_value <chr>
## $ ethnicity_source_concept_id <int>
The cdm object simplifies OMOP analysis by providing a consistent interface to the complex CDM structure.
6. Exploring the OMOP CDM
Basic Counts and Summaries
Start with overall statistics:
# Total number of patients
cdm$person |> count() |> collect()
## # A tibble: 1 × 1
## n
## <int>
## 1 0
# Number of observation periods
cdm$observation_period |> count() |> collect()
## # A tibble: 1 × 1
## n
## <int>
## 1 0
Demographic Summary
Analyze patient demographics:
demographics <- cdm$person |>
summarise(
total_patients = n(),
avg_age = mean(year_of_birth, na.rm = TRUE),
distinct_genders = n_distinct(gender_concept_id)
) |>
collect()
Exploring Clinical Events
Since the mock CDM has limited tables, let’s examine observation periods:
observation_summary <- cdm$observation_period |>
group_by(person_id) |>
summarise(
count = n(),
avg_duration = mean(observation_period_end_date - observation_period_start_date, na.rm = TRUE)
) |>
arrange(desc(count)) |>
collect()
For a full OMOP CDM, you would examine condition occurrences and drug exposures similarly.
Temporal Patterns
Analyze observation periods over time:
monthly_observations <- cdm$observation_period |>
mutate(month = floor_date(observation_period_start_date, "month")) |>
group_by(month) |>
summarise(observation_count = n()) |>
collect()
This EDA provides insights into data quality, completeness, and patterns in your OMOP database.
7. Identifying patient characteristics
Why Patient Characteristics Matter: In observational research, understanding patient characteristics (demographics, comorbidities, medication history) is crucial for confounding control and generalizability. These features help ensure study groups are comparable and results are interpretable.
Calculating Age at Observation Start
Join person and observation_period tables to find when patients were first observed:
age_at_observation <- cdm$observation_period |>
inner_join(cdm$person, by = "person_id") |>
group_by(person_id) |>
summarise(
first_observation = min(observation_period_start_date),
birth_year = first(year_of_birth)
) |>
mutate(age_at_observation = year(first_observation) - birth_year) |>
collect()
## Warning: There was 1 warning in `dplyr::summarise()`.
## ℹ In argument: `first_observation = min(observation_period_start_date)`.
## Caused by warning in `min.default()`:
## ! no non-missing arguments to min; returning Inf
Clinical Insight: Calculating age at first observation helps understand the age distribution of patients in your database, which is important for study design and generalizability.
Identifying Comorbidities
Find patients with observation periods (as a proxy for clinical activity):
observation_activity <- cdm$observation_period |>
group_by(person_id) |>
summarise(
observation_count = n(),
total_observation_days = sum(observation_period_end_date - observation_period_start_date)
) |>
collect()
Clinical Insight: Observation periods indicate the time patients are actively followed in the database, which affects the completeness of their clinical history.
Using CohortCharacteristics for Standardized Summaries
Instead of manual joins, use the CohortCharacteristics package for standardized, reproducible summaries. (Note: This requires a full OMOP CDM with clinical event tables.)
library(CohortCharacteristics)
# First create a cohort
cdm <- generateConceptCohortSet(
cdm = cdm,
name = "diabetes",
conceptSet = list("type_2_diabetes" = 201826),
end = "observation_period_end_date",
limit = "first"
)
# Summarize characteristics of the diabetes cohort
characteristics <- cdm$diabetes |>
summariseCharacteristics(
ageGroup = list(c(0, 17), c(18, 64), c(65, 999)),
gender = TRUE,
priorObservation = TRUE
) |>
collect()
Why CohortCharacteristics? This package provides standardized summaries that are consistent across studies, making results more comparable and reducing errors.
Creating a Patient Feature Dataset
Combine multiple characteristics:
patient_features <- cdm$person |>
left_join(
cdm$observation_period |>
group_by(person_id) |>
summarise(observation_count = n()),
by = "person_id"
) |>
mutate(
age = 2023 - year_of_birth, # Assuming current year
has_observations = observation_count > 0
) |>
collect()
These techniques allow you to create rich feature sets for analysis or modeling.
8. Adding cohorts to the CDM
library(CodelistGenerator)
library(CohortConstructor)
Creating Codelists with CodelistGenerator
Define clinical concepts (using available concepts in mock CDM):
# Get concepts for Gender
gender_codes <- getDescendants(cdm, 8507) # MALE concept
# Note: In a full OMOP CDM, you would use concept names or IDs for clinical conditions
Generating Cohorts with CohortConstructor
Create a diabetes cohort (requires full OMOP CDM):
cdm <- generateConceptCohortSet(
cdm = cdm,
name = "diabetes",
conceptSet = list("type_2_diabetes" = diabetes_codes),
end = "observation_period_end_date",
limit = "first"
)
Create a metformin user cohort (requires full OMOP CDM):
cdm <- generateConceptCohortSet(
cdm = cdm,
name = "metformin_users",
conceptSet = list("metformin" = metformin_codes),
end = "observation_period_end_date",
limit = "first"
)
Advanced Cohort Definitions
Combine multiple criteria:
# Patients with diabetes who started metformin within 1 year of diagnosis
cdm <- generateCohortSet(
cdm = cdm,
name = "diabetes_metformin",
cohortSet = data.frame(
cohort_definition_id = 1,
cohort_name = "Diabetes with Metformin",
# Define cohort entry criteria here
)
)
Validating Cohorts
Check cohort characteristics (requires cohort creation):
cohort_summary <- cdm$diabetes |>
summarise(
cohort_size = n(),
avg_index_age = mean(year(cohort_start_date) - year_of_birth, na.rm = TRUE)
) |>
collect()
These tools automate complex cohort creation, ensuring reproducibility and accuracy.
9. Working with cohorts
Characterizing Cohort Members
Join cohort with person data (requires cohort creation):
cohort_characteristics <- cdm$diabetes |>
inner_join(cdm$person, by = c("subject_id" = "person_id")) |>
summarise(
total_patients = n(),
avg_age = mean(2023 - year_of_birth, na.rm = TRUE),
distinct_genders = n_distinct(gender_concept_id)
) |>
collect()
Comparing Cohorts
Compare diabetes cohort to general population (requires cohort creation):
diabetes_vs_general <- bind_rows(
cdm$diabetes |>
inner_join(cdm$person, by = c("subject_id" = "person_id")) |>
mutate(group = "diabetes"),
cdm$person |>
anti_join(cdm$diabetes, by = c("person_id" = "subject_id")) |>
mutate(group = "general")
) |>
group_by(group) |>
summarise(avg_age = mean(2023 - year_of_birth, na.rm = TRUE)) |>
collect()
Analyzing Outcomes Over Time
Track outcomes after cohort entry (requires full OMOP CDM):
post_cohort_outcomes <- cdm$diabetes |>
left_join(cdm$condition_occurrence, by = c("subject_id" = "person_id")) |>
filter(condition_start_date >= cohort_start_date) |>
group_by(subject_id) |>
summarise(
follow_up_conditions = n(),
time_to_first_condition = min(condition_start_date - cohort_start_date, na.rm = TRUE)
) |>
collect()
Building Study Datasets
Create analysis-ready datasets (requires full OMOP CDM):
study_dataset <- cdm$diabetes |>
left_join(cdm$person, by = c("subject_id" = "person_id")) |>
left_join(
cdm$condition_occurrence |>
filter(condition_start_date <= cohort_start_date) |>
group_by(person_id) |>
summarise(pre_cohort_conditions = n()),
by = c("subject_id" = "person_id")
) |>
mutate(
age_at_entry = year(cohort_start_date) - year_of_birth,
has_comorbidities = pre_cohort_conditions > 0
) |>
select(subject_id, cohort_start_date, age_at_entry, gender_concept_id, has_comorbidities) |>
collect()
This workflow forms the foundation for comparative effectiveness research, population-level epidemiology, and patient-level prediction studies.
Glossary of Key Terms
- Cohort: A defined group of patients who meet specific inclusion/exclusion criteria for a study.
- Concept ID: A standardized numeric identifier for medical terms in the OMOP vocabulary.
- Domain: The category of a concept (e.g., Condition, Drug, Measurement).
- Index Date: The date that defines cohort entry (e.g., first diagnosis date).
- Incidence: The rate of new cases of a condition in a population over time.
- Prevalence: The proportion of a population with a condition at a specific point in time.
- Washout Period: A time period before cohort entry to ensure patients are “new” to treatment or condition.
- Vocabulary: A controlled set of terms used to standardize medical concepts across different data sources.
Core dplyr Verbs for Database Work
The tidyverse provides a consistent set of “verbs” for data manipulation that work seamlessly with databases through dbplyr.
| Purpose | Functions | Description |
|---|---|---|
| Selecting rows | filter(), distinct() | Select rows based on conditions. |
| Ordering rows | arrange() | Order rows by one or more columns. |
| Column Transformation | mutate(), select(), rename() | Create, modify, or select columns. |
| Grouping | group_by(), ungroup() | Group data for summarized calculations. |
| Aggregation | summarise(), count() | Calculate summary statistics. |
| Joining Tables | inner_join(), left_join(), etc. | Combine data from multiple tables. |
By mastering these verbs, you can construct powerful and readable data analysis pipelines that are executed directly in the database, ensuring scalability and performance.
Applying Tidyverse Principles to the OMOP CDM
While the principles of dbplyr and dplyr are powerful for any database, the OHDSI and DARWIN EU communities have developed a suite of R packages that build on this foundation to provide a seamless experience for working with the OMOP CDM.
The CDMConnector Package: Your Gateway to OMOP Data
The cornerstone of this ecosystem is the CDMConnector package. It allows you to create a cdm object, which is a special type of database connection that understands the structure of the OMOP CDM.
library(CDMConnector)
library(duckdb)
# For this example, we'll use a mock dataset
cdm <- mockCdmReference()
The cdm object gives you easy access to all the OMOP tables as lazy tibbles, ready to be used with dplyr.
# Access the person table
cdm$person
## # A tibble: 0 × 18
## # ℹ 18 variables: person_id <int>, gender_concept_id <int>,
## # year_of_birth <int>, month_of_birth <int>, day_of_birth <int>,
## # birth_datetime <date>, race_concept_id <int>, ethnicity_concept_id <int>,
## # location_id <int>, provider_id <int>, care_site_id <int>,
## # person_source_value <chr>, gender_source_value <chr>,
## # gender_source_concept_id <int>, race_source_value <chr>,
## # race_source_concept_id <int>, ethnicity_source_value <chr>, …
compute() vs. collect(): Working in the Database
A key concept when working with database-backed data is the difference between collect() and compute().
collect(): This function pulls data out of the database and into an R data frame in your computer’s memory. You should only use this on small, aggregated result sets that you need for visualization or local modeling.compute(): This function executes the query steps you’ve defined and saves the result as a new table in the database. This is essential for multi-step analyses, allowing you to build intermediate datasets without ever leaving the database, which is far more efficient.
Defining Clinical Ideas with CodelistGenerator
In any clinical study, you work with concepts like “Type 2 Diabetes” or “Metformin”, not abstract concept IDs. The CodelistGenerator package is used to gather all the relevant concept IDs for a clinical idea into a single object.
library(CodelistGenerator)
# Get all concepts for a gender concept and its descendants (example)
gender_codes <- getDescendants(cdm, 8507) # MALE concept
A Realistic Cohort with CohortConstructor
Now, let’s tackle a real-world problem: creating a cohort of patients with a first-time diagnosis of Type 2 Diabetes. This is where the power of the OHDSI tools becomes clear.
This task requires us to:
- Find all diabetes diagnoses in the
condition_occurrencetable using ourdiabetes_codes. - For each person, identify their very first diagnosis date.
- Create a cohort table with the person ID, the index date (first diagnosis), and the cohort definition ID.
The CohortConstructor package streamlines this. Here’s how you would do it:
library(CohortConstructor)
cdm <- generateConceptCohortSet(
cdm = cdm,
name = "diabetes",
conceptSet = list("type_2_diabetes" = diabetes_codes),
end = "observation_period_end_date",
limit = "first"
)
This single command performs all the necessary filtering, grouping, and joining on the database side to create a new cohort table, cdm$diabetes.
Answering a Clinical Question: Joining Cohorts and Data
Now that we have our cohort, we can start asking questions. For example: “What is the age and gender distribution of our new diabetes cohort?”
This requires joining our diabetes cohort with the person table. Because both are tables within the cdm object, this is a straightforward dplyr join.
diabetes_cohort <- cdm$diabetes
person_table <- cdm$person
cohort_demographics <- diabetes_cohort |>
inner_join(person_table, by = c("subject_id" = "person_id")) |>
select("subject_id", "cohort_start_date", "gender_concept_id", "year_of_birth") |>
mutate(age_at_diagnosis = year(cohort_start_date) - year_of_birth) |>
collect() # We collect here because the result is small and we want to analyze it in R
summary(cohort_demographics$age_at_diagnosis)
table(cohort_demographics$gender_concept_id)
This workflow—defining concepts, generating cohorts, and then using standard dplyr verbs to analyze the results—is the foundation of a powerful, scalable, and reproducible analysis pipeline in OHDSI.